La IA de DeepMind predice estructuras para un vasto tesoro de proteínas
En la imagen: El complejo de mediadores humanos ha sido durante mucho tiempo uno de los sistemas de proteínas múltiples más difíciles de entender para los biólogos estructurales. Crédito: Yuan He
El genoma humano contiene las instrucciones de más de 20.000 proteínas. Pero solo alrededor de un tercio de ellos han tenido sus estructuras 3D determinadas experimentalmente. Y en muchos casos, esas estructuras solo se conocen parcialmente.
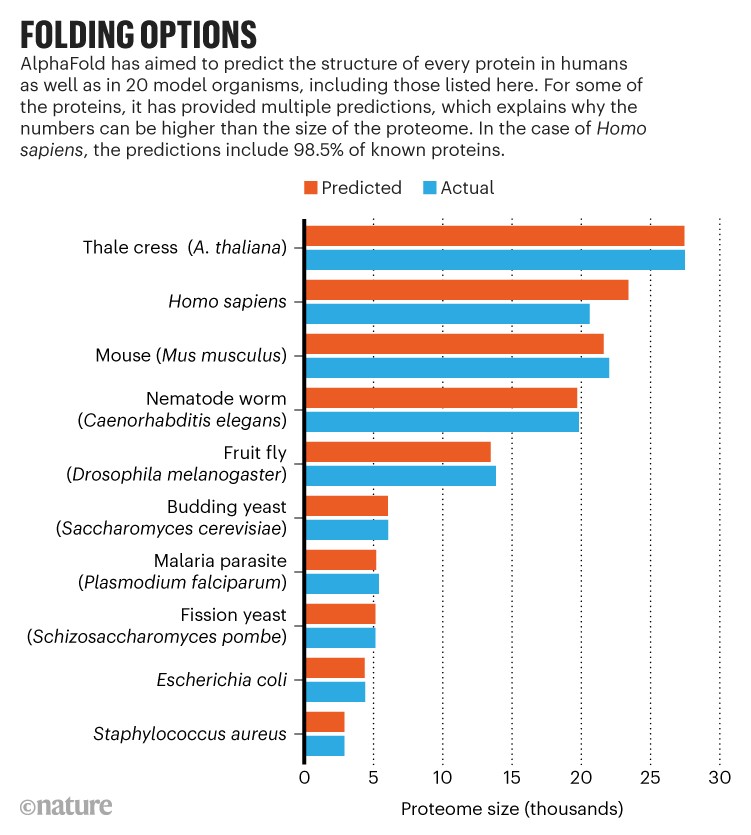
Ahora, una herramienta transformadora de inteligencia artificial (IA) llamada AlphaFold, que ha sido desarrollada por la empresa hermana de Google DeepMind en Londres, ha predicho la estructura de casi todo el proteoma humano (el complemento completo de proteínas expresadas por un organismo). Además, la herramienta ha predicho proteomas casi completos para varios otros organismos, desde ratones y maíz (maíz) hasta el parásito de la malaria (consulte 'Opciones de plegado').
Las más de 350.000 estructuras de proteínas, que están disponibles a través de una base de datos pública, varían en su precisión. Pero los investigadores dicen que el recurso, que aumentará a 130 millones de estructuras para fin de año, tiene el potencial de revolucionar las ciencias de la vida.
Fuente: EMBL – EBI y https://swissmodel.expasy.org/repository
“Es totalmente transformador desde mi perspectiva. Tener las formas de todas estas proteínas realmente te da una idea de sus mecanismos ”, dice Christine Orengo, bióloga computacional del University College London (UCL).
“Esta es la mayor contribución que ha hecho un sistema de inteligencia artificial hasta ahora para promover el conocimiento científico. No creo que sea exagerado decir eso ”, dice Demis Hassabis, cofundador y director ejecutivo de DeepMind.
Pero los investigadores enfatizan que el volcado de datos es un comienzo, no un final. Querrán validar las predicciones y, lo que es más importante, aplicarlas a experimentos que hasta ahora eran imposibles. "Es un primer paso asombroso, que tengamos todos estos datos en esa escala", dice David Jones, un biólogo computacional de UCL que asesoró a DeepMind en una versión anterior de AlphaFold.
Predicciones premiadas
DeepMind sorprendió a la comunidad de las ciencias de la vida el año pasado, cuando una versión actualizada de AlphaFold barrió un ejercicio bienal de predicción de proteínas llamado CASP (Evaluación crítica de la predicción de la estructura de las proteínas). En esta competencia de larga duración, que tradicionalmente ha sido el dominio de los académicos, los investigadores predicen las estructuras de proteínas cuyas estructuras se han resuelto experimentalmente, pero aún no se han hecho públicas.
Algunas de las predicciones de AlphaFold estaban a la par con muy buenos modelos experimentales, y algunos científicos dijeron que la influencia de la red sería trascendental. La semana pasada, DeepMind lanzó el código fuente detrás de la última versión de AlphaFold y una descripción detallada de cómo se desarrolló 1 (los equipos académicos ya han comenzado a usar estos recursos para hacer predicciones útiles). En el proceso de preparación del código de AlphaFold para su lanzamiento público, DeepMind lo refinó para que el código se ejecute de manera más eficiente. Algunas de las predicciones de CASP tardaron días, pero la versión actualizada de AlphaFold ahora podía calcularlas en minutos u horas.
Con esta eficiencia adicional, el equipo de DeepMind se propuso predecir las estructuras de casi todas las proteínas conocidas codificadas por el genoma humano, así como las de 20 organismos modelo. Las estructuras están disponibles en una base de datos mantenida por EMBL-EBI (el Instituto Europeo de Bioinformática del Laboratorio Europeo de Biología Molecular) en Hinxton, Reino Unido.
Además de las estructuras predichas, que cubren el 98,5% de las proteínas humanas conocidas y un porcentaje similar para otros organismos, AlphaFold generó una medida de la confianza de sus predicciones. “Queremos darles a los experimentadores y biólogos una señal realmente clara de en qué partes de las predicciones deben confiar”, dice Kathryn Tunyasuvunakool, ingeniera científica de DeepMind y primera autora de un artículo de Nature que describe las predicciones del proteoma humano 2. Para el proteoma humano, el 58% de sus predicciones para las ubicaciones de los aminoácidos individuales fueron lo suficientemente buenas como para confiar en la forma de los pliegues de la proteína, dice Tunyasuvunakool. Un subconjunto de esas predicciones, el 36% del total, son potencialmente lo suficientemente precisas como para detallar las características atómicas útiles para el diseño de fármacos, como el sitio activo de una enzima.
Incluso las predicciones menos precisas pueden ofrecer información. Los biólogos piensan que una gran proporción de proteínas humanas y las de otros eucariotas (organismos con células que tienen núcleos) contienen regiones que están intrínsecamente desordenadas y adquieren una estructura definida solo en concierto con otras moléculas. “Muchas proteínas simplemente se mueven en una solución, no tienen una estructura fija”, dice el investigador principal de AlphaFold, John Jumper. Algunas de las regiones que AlphaFold predijo con poca confianza coinciden con las que los biólogos sospechan que están desordenadas, dice Pushmeet Kohli, jefe de inteligencia artificial para la ciencia en DeepMind.
Determinar cómo las proteínas individuales interactúan con otros jugadores celulares es uno de los mayores desafíos para las predicciones de AlphaFold, dicen los investigadores. Para la competencia CASP, la mayoría de sus predicciones fueron de unidades de plegamiento independientes de una proteína, llamadas dominios. Pero el proteoma humano, y los de otros organismos, contiene proteínas con múltiples dominios que se pliegan de forma semiindependiente. Las células humanas también contienen moléculas compuestas por múltiples cadenas de proteínas que interactúan, como receptores en las membranas celulares.
Diluvio de datos
Las aproximadamente 365.000 predicciones de estructura depositadas esta semana deberían aumentar a 130 millones, casi la mitad de todas las proteínas conocidas, para fin de año, dice Sameer Velankar, bioinformático estructural en EMBL-EBI. La base de datos se actualizará a medida que se identifiquen nuevas proteínas y se mejoren las predicciones. “Este no es un recurso al que esperas tener acceso”, dice Tunyasuvunakool, y está ansiosa por ver qué se les ocurre a los científicos.
Los investigadores ya están utilizando AlphaFold y herramientas relacionadas para ayudar a entender los datos experimentales generados mediante cristalografía de rayos X y microscopía crioelectrónica. Marcelo Sousa, bioquímico de la Universidad de Colorado Boulder, usó AlphaFold para hacer modelos a partir de datos de rayos X de proteínas que las bacterias usan para evadir un antibiótico llamado colistina. Las partes del modelo experimental que diferían de la predicción de AlphaFold eran típicamente regiones que el software había asignado con poca confianza, señala Sousa, una señal de que AlphaFold está prediciendo con precisión sus límites.
Aún así, los biólogos querrán continuar comparando estas predicciones con datos experimentales para manejar mejor su confiabilidad, dice Venki Ramakrishnan, biólogo estructural del Laboratorio de Biología Molecular MRC en Cambridge, Reino Unido. “Necesitamos poder confiar en estos datos”, agrega Orengo.
Jones está impresionado con lo que ha logrado la red. Pero dice que muchos de los modelos predichos por AlphaFold podrían haber sido generados con software anterior desarrollado por académicos. "Para la mayoría de las proteínas, esos resultados probablemente sean lo suficientemente buenos para muchas de las cosas que desea hacer". Los científicos decididos a obtener la estructura de cualquier proteína en particular probablemente podrían tener éxito utilizando enfoques experimentales.
Pero es probable que la disponibilidad de tantas estructuras de proteínas marque un “cambio de paradigma” en biología, dice Mohammed AlQuraishi, biólogo computacional de la Universidad de Columbia en la ciudad de Nueva York que trabaja en la predicción de estructuras de proteínas. Su campo ha gastado tanto tiempo y energía en predecir estructuras proteicas precisas a esta escala que aún no ha descubierto qué hacer con tales recursos. "Todo lo que hacemos hoy que se basa en una secuencia de proteínas, ahora podemos hacerlo con la estructura de las proteínas".
Orengo espera que la base de datos le ayude a comprender mejor las limitaciones estructurales de las proteínas. Ella ha mapeado una base de datos de proteínas conocidas en aproximadamente 5,000 'familias estructurales', pero aproximadamente la mitad de las proteínas en la base de datos están excluidas porque no hay nada como ellos para el cual se ha determinado una estructura. Las predicciones de AlphaFold podrían ayudar a descubrir nuevas formas, dice. "Realmente veremos cómo se ve el espacio plegable".
Jones espera que AlphaFold lleve a un gran examen de conciencia entre los biólogos sobre qué hacer con tantas estructuras y la facilidad de crear muchas más. “Habrá conferencias. Ahora que tenemos 130 millones de modelos, ¿cómo cambia esto nuestra visión de la biología? Puede ser que no lo cambie ”, dice. "Sospecho que lo hará".
Publicación:22/Julio/2021
Fuente:
Revista científica Nature
Tags de búsqueda:DeepMind, Inteligencia Artificial, Google, Biología, Biología molecular, ADN, Genética, Salud, Medicina, Investigación, Ciencia, Actualidad
 (57)+604 4480388
(57)+604 4480388